RNA Sequencing
You can find this application in the demos folder of your Jupyter notebook environment.
- 📝 samplesheet.csv
- 📝 rnaseq_workflow.ipynb
RNA sequencing (RNA-seq) is a key technique in modern biology, used to quantify gene expression, detect alternative splicing, and understand transcriptional changes under different conditions—whether in development, disease, or response to treatment. This notebook demonstrates a full RNA-seq analysis workflow powered by Nextflow and the nf-core/rnaseq pipeline, showcasing how Camber simplifies and scales reproducible cloud-based analysis.
The first step is to import the nextflow package:
from camber import nextflowHere’s an example of how to setup configurations and execute a job:
command: The full Nextflow command to run the nf-core/rnaseq pipeline.--input:"./samplesheet.csv": the relative path ofsamplesheet.csvfile to the current notebook. In case of using local fastq files, the locations insamplesheet.csvfile content are relative.--outdir:"./outputs": the location stores output data of the job.
node_size="MICRO": indicate node size to perform the job.num_nodes=4: indicate number of nodes to run workflow tasks in parallel when possible.
command = "nextflow run nf-core/rnaseq \
--aligner star_rsem \
--fasta s3://camber-open-storage-prod/public/fastq/rnaseq/ITAG2.3_genomic_Ch6.fasta \
--gtf s3://camber-open-storage-prod/public/fastq/rnaseq/ITAG_pre2.3_gene_models_Ch6.gtf \
--input ./samplesheet.csv \
--outdir ./outputs \
--skip_biotype_qc true \
-r 3.18.0"nf_rnaseq_job = nextflow.create_job(
command=command,
node_size="XXSMALL",
num_nodes=4
)This step is to check job status:
nf_rnaseq_job.statusView job logs online:
nf_rnaseq_job.read_logs()When the job is done, you can discover and download the results of the job by two ways:
- Browser data directly in notebook environment:
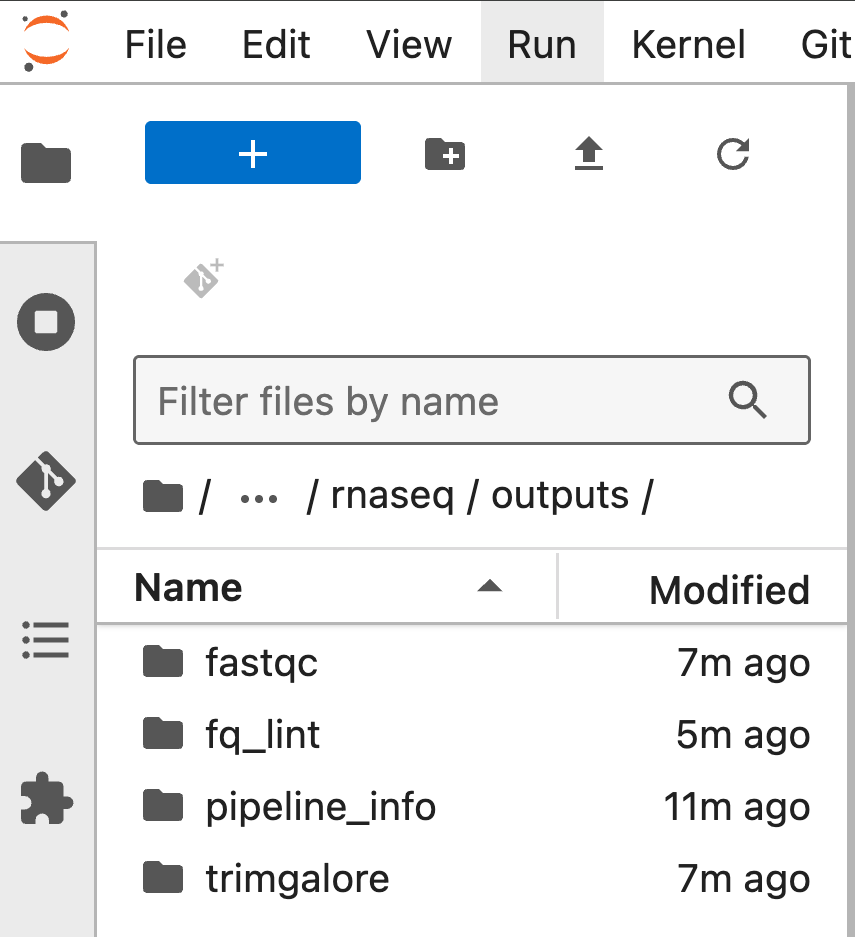
- Go to the Stash UI:
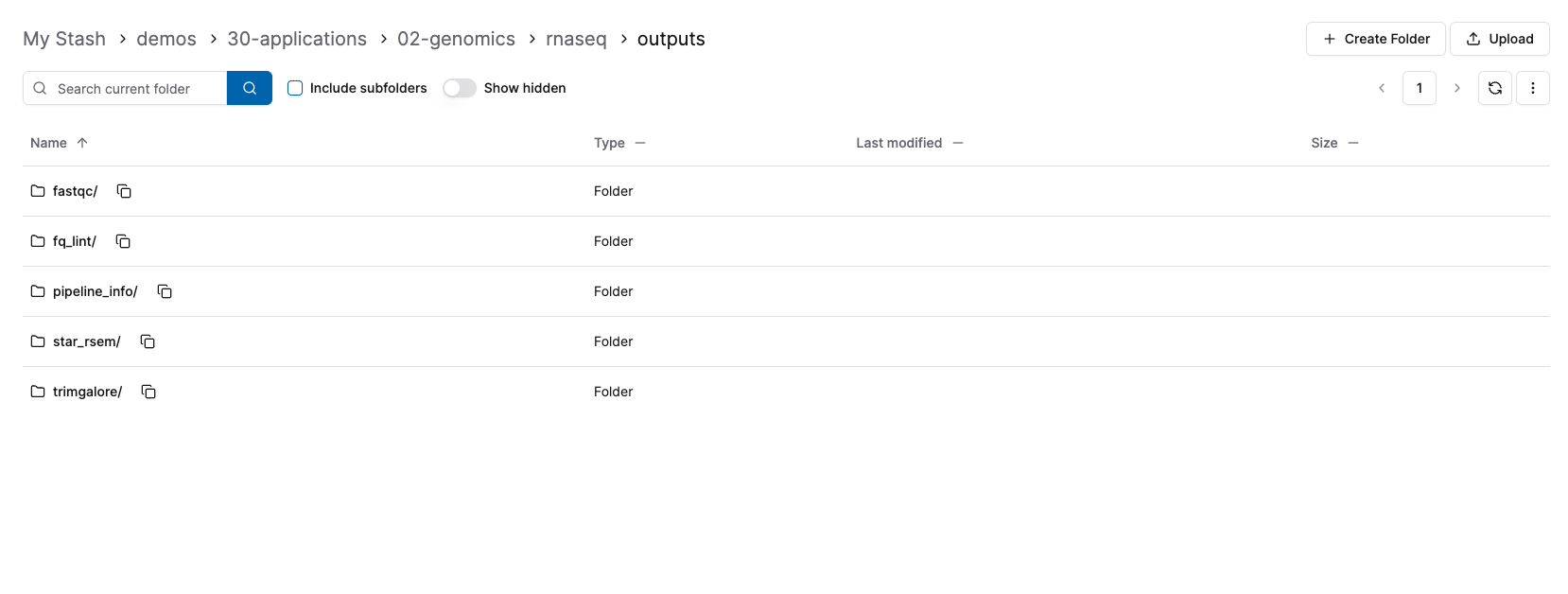
By running this RNA-seq pipeline on Camber, you’ve leveraged a reproducible, cloud-optimized workflow with minimal infrastructure overhead. This approach streamlines large-scale data analysis and sets the stage for scalable genomics research using community standards and modern tools.
Note: Please note that the files and folders saved in the demos directory are temporary and will be reset after each JupyterHub session. We recommend changing the value of --outdir to a different location if you wish to store your data permanently.